Generate multi-omics data
Assume that you’ve complete the model training process. You can run the following command to generate new multiome latent representations:
cd script/training_diffusion
sh ssh_scripts/multimodal_sample.sh
Change the MULTIMODAL_MODEL_PATH in the sh file to the checkpoint path in step 2, and the DATA_DIR to your local data path. Also change other parameters to align with the training process. The specific_type is to specify the generation cell type. If not set, generate cell in proportion to the cell type in the original dataset by default. The all_save_num is the number of cells you want to generate.
After that, the generated cells latent representations will be saved in the OUT_DIR. You can follow the below script to decode them back to original space.
First thing first, import packages and set related paths:
[1]:
import os
os.environ['CUDA_VISIBLE_DEVICES'] = '4'
import scanpy as sc
import anndata as ad
import numpy as np
import torch
import torch.nn.functional as F
import matplotlib.pyplot as plt
from scipy import stats
from scdiffusionX.utils import MMD, LISI, random_forest, norm_total
import pandas as pd
from torch.distributions import Normal
from scvi.distributions import NegativeBinomial
from torch.distributions import Poisson, Bernoulli
import muon as mu
import yaml
import seaborn as sns
from tqdm import tqdm
from scdiffusionX.Autoencoder.data.scrnaseq_loader import RNAseqLoader
from scdiffusionX.Autoencoder.models.base.encoder_model import EncoderModel
/home/lep/miniconda3/envs/test/lib/python3.8/site-packages/lightning_lite/__init__.py:29: DeprecationWarning: Deprecated call to `pkg_resources.declare_namespace('lightning_lite')`.
Implementing implicit namespace packages (as specified in PEP 420) is preferred to `pkg_resources.declare_namespace`. See https://setuptools.pypa.io/en/latest/references/keywords.html#keyword-namespace-packages
__import__("pkg_resources").declare_namespace(__name__)
/home/lep/miniconda3/envs/test/lib/python3.8/site-packages/pytorch_lightning/__init__.py:45: DeprecationWarning: Deprecated call to `pkg_resources.declare_namespace('pytorch_lightning')`.
Implementing implicit namespace packages (as specified in PEP 420) is preferred to `pkg_resources.declare_namespace`. See https://setuptools.pypa.io/en/latest/references/keywords.html#keyword-namespace-packages
__import__("pkg_resources").declare_namespace(__name__)
/home/lep/miniconda3/envs/test/lib/python3.8/site-packages/umap/__init__.py:9: ImportWarning: Tensorflow not installed; ParametricUMAP will be unavailable
warn(
[9]:
# set model and data path
encoder_config = "/stor/lep/workspace/scDiffusion-X/script/training_autoencoder/configs/encoder/encoder_multimodal.yaml"
dataset_path = '/stor/lep/diffusion/multiome/openproblem_filtered.h5mu'
covariate_keys = "cell_type" #"leiden"
num_class = 22
ae_path = "/stor/lep/workspace/scDiffusion-X/Autoencoder/project_folder/experiments/train_autoencoder_openproblem_multimodal/checkpoints/last.ckpt"
Then, load the dataset:
[3]:
mdata = mu.read_h5mu(dataset_path)
gene_names = mdata['rna'].var_names
real_cell = mdata['rna'][::10].X.toarray()
real_cell2 = mdata['atac'][::10].X.toarray()
cell_type = mdata['rna'][::10].obs['cell_type'].values
type_list = np.unique(mdata['rna'][::10].obs['cell_type'].values)
real_cell.shape
/home/lep/miniconda3/envs/test/lib/python3.8/site-packages/anndata/_core/anndata.py:453: PendingDeprecationWarning: The dtype argument will be deprecated in anndata 0.10.0
warnings.warn(
/home/lep/miniconda3/envs/test/lib/python3.8/site-packages/anndata/_core/anndata.py:453: PendingDeprecationWarning: The dtype argument will be deprecated in anndata 0.10.0
warnings.warn(
[3]:
(6925, 13431)
After that, decode the generated latent representations back to original space:
[7]:
# get size factor for encoder
dataset = RNAseqLoader(data_path=dataset_path,
layer_key='X_counts',
covariate_keys=[covariate_keys],
subsample_frac=1,
encoder_type='learnt_autoencoder',
multimodal=True,
is_binarized=True)
size_factor_statistics = {"mean": dataset.log_size_factor_mu,
"sd": dataset.log_size_factor_sd}
def get_size_factor(type_index):
covariate_indices = {}
covariate_indices[covariate_keys] = type_index
mean_size_factor, sd_size_factor = size_factor_statistics["mean"][covariate_keys], size_factor_statistics["sd"][covariate_keys]
mean_size_factor, sd_size_factor = mean_size_factor[covariate_indices[covariate_keys]], sd_size_factor[covariate_indices[covariate_keys]]
size_factor_dist = Normal(loc=mean_size_factor, scale=sd_size_factor)
log_size_factor = size_factor_dist.sample().view(-1, 1)
size_factor = torch.exp(log_size_factor)
return {"rna": size_factor}
/home/lep/miniconda3/envs/test/lib/python3.8/site-packages/anndata/_core/anndata.py:453: PendingDeprecationWarning: The dtype argument will be deprecated in anndata 0.10.0
warnings.warn(
/home/lep/miniconda3/envs/test/lib/python3.8/site-packages/anndata/_core/anndata.py:453: PendingDeprecationWarning: The dtype argument will be deprecated in anndata 0.10.0
warnings.warn(
[10]:
# Load encoder and decoder
with open(encoder_config, 'r') as file:
yaml_content = file.read()
autoencoder_args = yaml.safe_load(yaml_content)
# Initialize encoder
autoencoder_args['encoder_kwargs']['rna']['norm_type']='batchnorm'
autoencoder_args['encoder_kwargs']['atac']['norm_type']='batchnorm'
encoder_model = EncoderModel(in_dim={'atac': real_cell2.shape[1], 'rna': real_cell.shape[1]},
n_cat=mdata['rna'].obs[covariate_keys].unique().shape[0],
conditioning_covariate=covariate_keys,
encoder_type='learnt_autoencoder',
**autoencoder_args)
# Load weights
encoder_model.load_state_dict(torch.load(ae_path)["state_dict"])
/tmp/ipykernel_2162493/801034232.py:16: FutureWarning: You are using `torch.load` with `weights_only=False` (the current default value), which uses the default pickle module implicitly. It is possible to construct malicious pickle data which will execute arbitrary code during unpickling (See https://github.com/pytorch/pytorch/blob/main/SECURITY.md#untrusted-models for more details). In a future release, the default value for `weights_only` will be flipped to `True`. This limits the functions that could be executed during unpickling. Arbitrary objects will no longer be allowed to be loaded via this mode unless they are explicitly allowlisted by the user via `torch.serialization.add_safe_globals`. We recommend you start setting `weights_only=True` for any use case where you don't have full control of the loaded file. Please open an issue on GitHub for any issues related to this experimental feature.
encoder_model.load_state_dict(torch.load(ae_path)["state_dict"])
[10]:
<All keys matched successfully>
Note: these data (rna_seq, atac_seq, and type_index) are saved in your OUT_DIR, change the path to your own.
[12]:
# read generated latent representation and decode back to gene expression/atac seq
rna_seq = np.load('../outputs/samples_new/w512_scale124_drop0_80w_rescale10_3crossatt64_condi/dpm_solver_80w_condi_big/RNA_0.npz')['data']
atac_seq = np.load('../outputs/samples_new/w512_scale124_drop0_80w_rescale10_3crossatt64_condi/dpm_solver_80w_condi_big/ATAC_0.npz')['data']
type_index = np.load('../outputs/samples_new/w512_scale124_drop0_80w_rescale10_3crossatt64_condi/dpm_solver_80w_condi_big/RNA_0.npz')['label']
# load norm factor for encoder
npzfile = np.load('/'.join(ae_path.split('/')[:-2])+'/norm_factor.npz')
rna_std = npzfile['rna_std']
atac_std = npzfile['atac_std']
z = {'rna':torch.tensor(rna_seq*rna_std).squeeze(1),'atac':torch.tensor(atac_seq*atac_std).squeeze(1)}
# get size factor and decode
size_factor = get_size_factor(torch.tensor(type_index,dtype=torch.int))
mu_hat = encoder_model.decode(z, size_factor)
sample = {} # containing final samples
for mod in mu_hat:
if mod=="rna":
distr = NegativeBinomial(mu=mu_hat[mod], theta=torch.exp(encoder_model.theta))
else: # if mod is atac
if not encoder_model.is_binarized:
distr = Poisson(rate=mu_hat[mod])
else:
distr = Bernoulli(probs=mu_hat[mod])
sample[mod] = distr.sample()
reconstruct = sample['rna'][:real_cell.shape[0]].detach().numpy()
reconstruct2 = sample['atac'][:real_cell.shape[0]].detach().numpy()
type_index = type_index[:real_cell.shape[0]]
reconstruct.shape
[12]:
(6925, 13431)
Here reconstruct is the generated scRNA-seq, reconstruct2 is the gerated scATAC-seq data. You can evaluate your generation results with the following scripts:
[7]:
print('RNA')
print(real_cell.mean())
print(reconstruct.mean())
print('spearman=',stats.spearmanr(real_cell.mean(axis=0), reconstruct.mean(axis=0)).correlation)
print('pearson=',np.corrcoef(real_cell.mean(axis=0), reconstruct.mean(axis=0))[0][1])
print('MSE=',np.mean((reconstruct-real_cell)**2))
print('L2 Norm=',np.linalg.norm(reconstruct.mean(axis=0)-real_cell.mean(axis=0)))
print('\nATAC')
print(real_cell2.mean())
print(reconstruct2.mean())
print('spearman=',stats.spearmanr(real_cell2.mean(axis=0), reconstruct2.mean(axis=0)).correlation)
print('pearson=',np.corrcoef(real_cell2.mean(axis=0), reconstruct2.mean(axis=0))[0][1])
print('MSE=',np.mean((reconstruct2-real_cell2)**2))
print('L2 Norm=',np.linalg.norm(reconstruct2.mean(axis=0)-real_cell2.mean(axis=0)))
RNA
0.14379396
0.14475352
spearman= 0.9913733776473627
pearson= 0.987588422541502
MSE= 7.6237383
L2 Norm= 13.169576
ATAC
0.07920893
0.07917095
spearman= 0.9850661563393119
pearson= 0.9977483111114237
MSE= 0.13435104
L2 Norm= 0.97393376
[8]:
print('RNA')
adata = np.concatenate((real_cell, reconstruct),axis=0)
adata = ad.AnnData(adata, dtype=np.float32)
adata.obs_names = [f"true_Cell" for i in range(real_cell.shape[0])]+[f"gen_Cell" for i in range(reconstruct.shape[0])]
sc.tl.pca(adata, svd_solver='arpack')
adata.obs['batch'] = pd.Categorical([f"true_Cell" for i in range(real_cell.shape[0])]+[f"gen_Cell" for i in range(reconstruct.shape[0])])
print('MMD = ', MMD(adata))
print('LISI = ', LISI(adata))
print('AUC = ', random_forest(adata))
print('\nATAC')
adata = np.concatenate((real_cell2, reconstruct2),axis=0)
adata = ad.AnnData(adata, dtype=np.float32)
adata.obs_names = [f"true_Cell" for i in range(real_cell2.shape[0])]+[f"gen_Cell" for i in range(reconstruct2.shape[0])]
sc.tl.pca(adata, svd_solver='arpack')
adata.obs['batch'] = pd.Categorical([f"true_Cell" for i in range(real_cell2.shape[0])]+[f"gen_Cell" for i in range(reconstruct2.shape[0])])
print('MMD = ', MMD(adata))
print('LISI = ', LISI(adata))
print('AUC = ', random_forest(adata))
RNA
100%|██████████| 70/70 [00:16<00:00, 4.31it/s]
MMD = tensor(0.0025)
LISI = 0.8510099970259501
AUC = 0.6675026483686849
ATAC
100%|██████████| 70/70 [00:15<00:00, 4.44it/s]
MMD = tensor(0.0008)
LISI = 0.9244179941358288
AUC = 0.5611923262683485
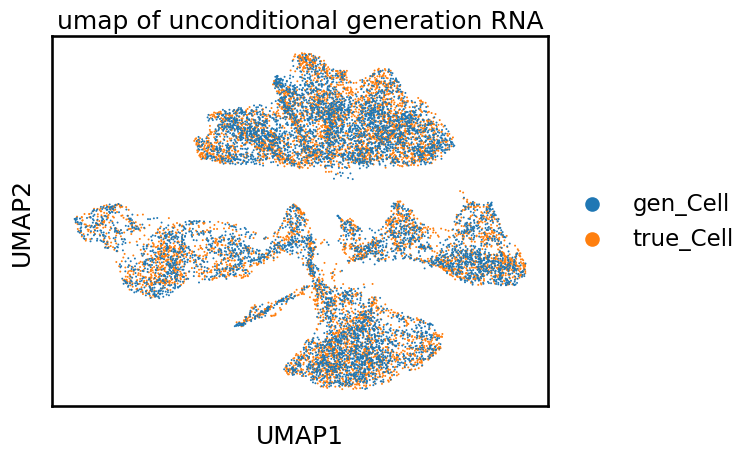
You can also draw the UMAP to see if the generated data align well with the original data:
[ ]:
adata = np.concatenate((real_cell, reconstruct),axis=0)
adata = ad.AnnData(adata, dtype=np.float32)
adata.obs['cell_name'] = [f"true_Cell" for i in range(real_cell.shape[0])]+[f"gen_Cell" for i in range(reconstruct.shape[0])]
adata.obs['cell_type'] = list(cell_type) + ['gen '+type_list[type] for type in type_index]
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
adata.raw = adata
adata = adata[:, adata.var.highly_variable]
sc.pp.scale(adata)
sc.tl.pca(adata, svd_solver='arpack')
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=20)
sc.tl.umap(adata)
plt.rcParams['pdf.fonttype'] = 42
plt.rcParams['ps.fonttype'] = 42
sc.pl.umap(adata, color='cell_name', size=8, title='umap of unconditional generation RNA')
# plt.legend(loc = 'upper right')
[24]:
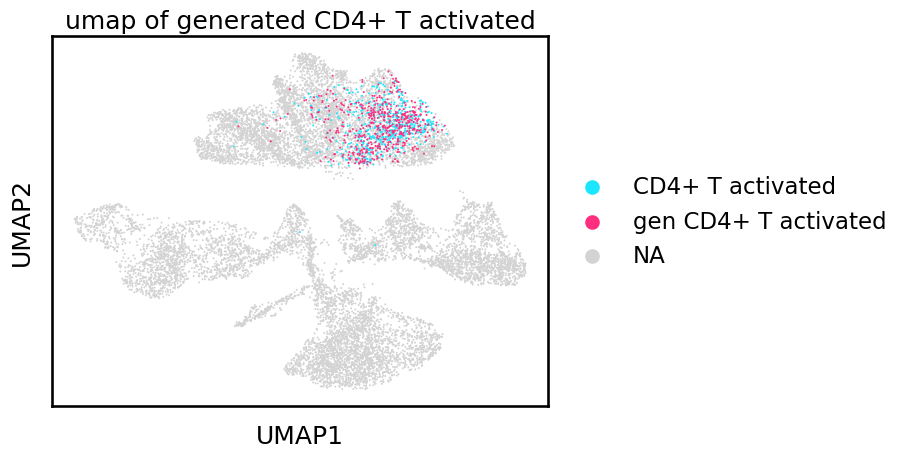
sc.pl.umap(adata, color='cell_type',groups=['CD4+ T activated','gen CD4+ T activated'], size=8, title='umap of generated CD4+ T activated')
# plt.legend(loc = 'upper right')
[16]:
adata = np.concatenate((real_cell2, reconstruct2),axis=0)
adata = ad.AnnData(adata, dtype=np.float32)
# adata.var_names = gene_names
adata.obs['cell_name'] = [f"true_Cell" for i in range(real_cell2.shape[0])]+[f"gen_Cell" for i in range(reconstruct2.shape[0])]
sc.pp.normalize_total(adata, target_sum=1e4)
# sc.pp.log1p(adata)
# sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# adata.raw = adata
# adata = adata[:, adata.var.highly_variable]
# sc.pp.scale(adata)
sc.tl.pca(adata, svd_solver='arpack')
sc.pp.neighbors(adata, n_neighbors=10, n_pcs=20)
sc.tl.umap(adata)
plt.rcParams['pdf.fonttype'] = 42
plt.rcParams['ps.fonttype'] = 42
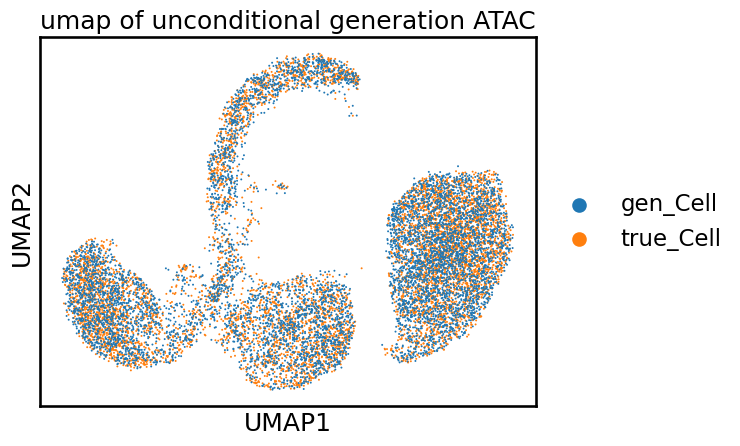
sc.pl.umap(adata, color='cell_name', size=8, title='umap of unconditional generation ATAC')
# plt.legend(loc = 'upper right')
/home/lep/miniconda3/envs/test/lib/python3.8/site-packages/anndata/_core/anndata.py:453: PendingDeprecationWarning: The dtype argument will be deprecated in anndata 0.10.0
warnings.warn(
/home/lep/miniconda3/envs/test/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:394: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
For each cell type, you can evaluate them one by one. Here is an example on the Openproblem dataset:
[ ]:
from tqdm import tqdm
type_list = ['B1 B', 'CD14+ Mono', 'CD16+ Mono', 'CD4+ T activated',
'CD4+ T naive', 'CD8+ T', 'CD8+ T naive', 'Erythroblast',
'G/M prog', 'HSC', 'ID2-hi myeloid prog', 'ILC', 'Lymph prog',
'MK/E prog', 'NK', 'Naive CD20+ B', 'Normoblast', 'Plasma cell',
'Proerythroblast', 'Transitional B', 'cDC2', 'pDC']
columns = ['cell type', 'RNA PCC', 'RNA SCC', 'RNA MMD', 'RNA LISI', 'RNA AUC', 'ATAC PCC', 'ATAC SCC', 'ATAC MMD', 'ATAC LISI', 'ATAC AUC']
type_index = np.load('../outputs/samples_new/w512_scale124_drop0_80w_rescale10_3crossatt64_condi/dpm_solver_80w_condi_big/RNA_0.npz')['label'] # here we use all sampled data
df = pd.DataFrame(columns=columns)
for id, type in enumerate(type_list):
# load different cell type and control the number of each type (to save evaluation time)
total = (mdata['rna'].obs['cell_type']==type).sum()
sample_index = np.random.choice(total, size=800, replace=False) if total>=800 else list(range(total))
real_cell = mdata['rna'][mdata['rna'].obs['cell_type']==type].X.toarray()[sample_index]
real_cell2 = mdata['atac'][mdata['atac'].obs['cell_type']==type].X.toarray()[sample_index]
cell_num = min((type_index==id).sum(),real_cell.shape[0])
reconstruct = sample['rna'][type_index==id][:cell_num].detach().numpy()
reconstruct2 = sample['atac'][type_index==id][:cell_num].detach().numpy()
real_cell = real_cell[:cell_num]
real_cell2 = real_cell2[:cell_num]
# calculate evaluation metric
rna_scc = stats.spearmanr(real_cell.mean(axis=0), reconstruct.mean(axis=0)).correlation
rna_pcc = np.corrcoef(real_cell.mean(axis=0), reconstruct.mean(axis=0))[0][1]
atac_scc = stats.spearmanr(real_cell2.mean(axis=0), reconstruct2.mean(axis=0)).correlation
atac_pcc = np.corrcoef(real_cell2.mean(axis=0), reconstruct2.mean(axis=0))[0][1]
adata = np.concatenate((real_cell, reconstruct),axis=0)
adata = ad.AnnData(adata, dtype=np.float32)
adata.obs_names = [f"true_Cell" for i in range(real_cell.shape[0])]+[f"gen_Cell" for i in range(reconstruct.shape[0])]
sc.tl.pca(adata, svd_solver='arpack')
adata.obs['batch'] = pd.Categorical([f"true_Cell" for i in range(real_cell.shape[0])]+[f"gen_Cell" for i in range(reconstruct.shape[0])])
rna_mmd = MMD(adata)
rna_lisi = LISI(adata)
rna_auc = random_forest(adata)
adata = np.concatenate((real_cell2, reconstruct2),axis=0)
adata = ad.AnnData(adata, dtype=np.float32)
adata.obs_names = [f"true_Cell" for i in range(real_cell2.shape[0])]+[f"gen_Cell" for i in range(reconstruct2.shape[0])]
sc.tl.pca(adata, svd_solver='arpack')
adata.obs['batch'] = pd.Categorical([f"true_Cell" for i in range(real_cell2.shape[0])]+[f"gen_Cell" for i in range(reconstruct2.shape[0])])
atac_mmd = MMD(adata)
atac_lisi = LISI(adata)
atac_auc = random_forest(adata)
df.loc[id] = [type, rna_pcc, rna_scc, rna_mmd, rna_lisi, rna_auc, atac_pcc, atac_scc, atac_mmd, atac_lisi, atac_auc]
100%|██████████| 9/9 [00:00<00:00, 69.95it/s]
100%|██████████| 9/9 [00:00<00:00, 69.71it/s]
100%|██████████| 9/9 [00:00<00:00, 70.13it/s]
100%|██████████| 9/9 [00:00<00:00, 92.92it/s]
100%|██████████| 9/9 [00:00<00:00, 90.47it/s]
100%|██████████| 9/9 [00:00<00:00, 92.73it/s]
100%|██████████| 9/9 [00:00<00:00, 98.30it/s]
100%|██████████| 9/9 [00:00<00:00, 97.56it/s]
100%|██████████| 9/9 [00:00<00:00, 98.03it/s]
100%|██████████| 9/9 [00:00<00:00, 93.87it/s]
100%|██████████| 9/9 [00:00<00:00, 95.36it/s]
100%|██████████| 9/9 [00:00<00:00, 74.83it/s]
100%|██████████| 9/9 [00:00<00:00, 68.71it/s]
100%|██████████| 9/9 [00:00<00:00, 47.93it/s]
100%|██████████| 9/9 [00:00<00:00, 78.03it/s]
100%|██████████| 9/9 [00:00<00:00, 91.08it/s]
100%|██████████| 9/9 [00:00<00:00, 100.39it/s]
100%|██████████| 9/9 [00:00<00:00, 99.14it/s]
100%|██████████| 9/9 [00:00<00:00, 93.89it/s]
100%|██████████| 9/9 [00:00<00:00, 90.95it/s]
100%|██████████| 1/1 [00:00<00:00, 448.35it/s]
100%|██████████| 1/1 [00:00<00:00, 504.97it/s]
100%|██████████| 8/8 [00:00<00:00, 90.17it/s]
100%|██████████| 8/8 [00:00<00:00, 92.64it/s]
100%|██████████| 9/9 [00:00<00:00, 96.32it/s]
100%|██████████| 9/9 [00:00<00:00, 99.24it/s]
100%|██████████| 9/9 [00:00<00:00, 102.57it/s]
100%|██████████| 9/9 [00:00<00:00, 96.32it/s]
100%|██████████| 9/9 [00:00<00:00, 100.60it/s]
100%|██████████| 9/9 [00:00<00:00, 83.09it/s]
100%|██████████| 9/9 [00:00<00:00, 95.19it/s]
100%|██████████| 9/9 [00:00<00:00, 94.20it/s]
100%|██████████| 9/9 [00:00<00:00, 95.01it/s]
100%|██████████| 9/9 [00:00<00:00, 93.78it/s]
100%|██████████| 4/4 [00:00<00:00, 139.75it/s]
100%|██████████| 4/4 [00:00<00:00, 85.28it/s]
100%|██████████| 9/9 [00:00<00:00, 61.82it/s]
100%|██████████| 9/9 [00:00<00:00, 47.60it/s]
100%|██████████| 9/9 [00:00<00:00, 77.95it/s]
100%|██████████| 9/9 [00:00<00:00, 73.52it/s]
100%|██████████| 8/8 [00:00<00:00, 86.21it/s]
100%|██████████| 8/8 [00:00<00:00, 83.80it/s]
100%|██████████| 9/9 [00:00<00:00, 74.63it/s]
100%|██████████| 9/9 [00:00<00:00, 88.48it/s]
[ ]:
df.to_csv('eval_results/diffusion.csv', index=False) # save it if you want
df
| cell type | RNA PCC | RNA SCC | RNA MMD | RNA LISI | RNA AUC | ATAC PCC | ATAC SCC | ATAC MMD | ATAC LISI | ATAC AUC | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | B1 B | 0.987566 | 0.967615 | tensor(0.0150) | 0.833364 | 0.797694 | 0.986721 | 0.979733 | tensor(0.0106) | 0.899214 | 0.595788 |
| 1 | CD14+ Mono | 0.992969 | 0.975501 | tensor(0.0269) | 0.873068 | 0.677646 | 0.981683 | 0.975874 | tensor(0.0077) | 0.905203 | 0.597564 |
| 2 | CD16+ Mono | 0.995610 | 0.982333 | tensor(0.0422) | 0.806425 | 0.786214 | 0.982685 | 0.974554 | tensor(0.0622) | 0.847424 | 0.660114 |
| 3 | CD4+ T activated | 0.997701 | 0.972190 | tensor(0.0183) | 0.879269 | 0.670718 | 0.988320 | 0.978231 | tensor(0.0072) | 0.888342 | 0.633578 |
| 4 | CD4+ T naive | 0.997946 | 0.974163 | tensor(0.0353) | 0.839855 | 0.745648 | 0.989293 | 0.979403 | tensor(0.0133) | 0.877567 | 0.604692 |
| 5 | CD8+ T | 0.996127 | 0.969788 | tensor(0.0249) | 0.784428 | 0.730742 | 0.987550 | 0.979525 | tensor(0.0238) | 0.911457 | 0.639081 |
| 6 | CD8+ T naive | 0.995426 | 0.973165 | tensor(0.0270) | 0.806572 | 0.658188 | 0.990184 | 0.981577 | tensor(0.0188) | 0.891647 | 0.647134 |
| 7 | Erythroblast | 0.998432 | 0.977005 | tensor(0.0042) | 0.922589 | 0.734019 | 0.989771 | 0.966028 | tensor(0.0187) | 0.886622 | 0.719638 |
| 8 | G/M prog | 0.981477 | 0.979162 | tensor(0.0316) | 0.823997 | 0.794243 | 0.981967 | 0.969076 | tensor(0.0131) | 0.892188 | 0.690201 |
| 9 | HSC | 0.987696 | 0.978155 | tensor(0.0364) | 0.797329 | 0.767057 | 0.983788 | 0.974396 | tensor(0.0166) | 0.853439 | 0.715586 |
| 10 | ID2-hi myeloid prog | 0.923833 | 0.735099 | tensor(0.4095) | 0.716665 | 0.837321 | 0.873203 | 0.788035 | tensor(0.0468) | 0.924559 | 0.488038 |
| 11 | ILC | 0.990462 | 0.960589 | tensor(0.0322) | 0.820974 | 0.752227 | 0.983203 | 0.964551 | tensor(0.0125) | 0.911292 | 0.629204 |
| 12 | Lymph prog | 0.981817 | 0.973881 | tensor(0.0234) | 0.857090 | 0.731117 | 0.986919 | 0.980634 | tensor(0.0418) | 0.869461 | 0.688000 |
| 13 | MK/E prog | 0.960135 | 0.977791 | tensor(0.0424) | 0.786968 | 0.773459 | 0.985934 | 0.977077 | tensor(0.0206) | 0.876386 | 0.718487 |
| 14 | NK | 0.994365 | 0.972740 | tensor(0.0311) | 0.768475 | 0.754477 | 0.987499 | 0.977424 | tensor(0.0165) | 0.885925 | 0.563350 |
| 15 | Naive CD20+ B | 0.993580 | 0.960503 | tensor(0.0211) | 0.830556 | 0.647759 | 0.985722 | 0.975846 | tensor(0.0122) | 0.894257 | 0.629877 |
| 16 | Normoblast | 0.997995 | 0.933665 | tensor(0.0201) | 0.905464 | 0.724090 | 0.985125 | 0.915431 | tensor(0.0624) | 0.840820 | 0.642357 |
| 17 | Plasma cell | 0.959082 | 0.961674 | tensor(0.0229) | 0.837189 | 0.833270 | 0.979633 | 0.963814 | tensor(0.0197) | 0.901915 | 0.604934 |
| 18 | Proerythroblast | 0.982554 | 0.984298 | tensor(0.0107) | 0.861554 | 0.727941 | 0.988878 | 0.981070 | tensor(0.0146) | 0.894323 | 0.668142 |
| 19 | Transitional B | 0.990861 | 0.954412 | tensor(0.0442) | 0.873715 | 0.750600 | 0.985322 | 0.980596 | tensor(0.0225) | 0.887747 | 0.643908 |
| 20 | cDC2 | 0.980364 | 0.979903 | tensor(0.0208) | 0.853975 | 0.712523 | 0.983508 | 0.973885 | tensor(0.0151) | 0.909913 | 0.697812 |
| 21 | pDC | 0.982402 | 0.980236 | tensor(0.0911) | 0.779413 | 0.822079 | 0.986729 | 0.976973 | tensor(0.0178) | 0.879067 | 0.674945 |