Decode gene regulation mechanism
This tutorial is to show how to use scDiffusion-X to resolve regulatory relationships between genes and chromatin regions (peaks). First, you need to finishi the training process on the dataset you are interested in. The following codes will show you how to obtain the cell-type specific heterogeneous gene regulatory network.
First, import the related packages.
[ ]:
import os
os.environ['CUDA_VISIBLE_DEVICES'] = '4'
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
import pandas as pd
from sklearn.cluster import KMeans
import torch
import torch.optim as optim
import muon as mu
import yaml
import os
from scdiffusionX.utils import *
from scdiffusionX.Autoencoder.data.scrnaseq_loader import RNAseqLoader
from scdiffusionX.Autoencoder.models.base.encoder_model import EncoderModel
import argparse
from scdiffusionX.DiffusionBackbone.multimodal_script_util import (
model_and_diffusion_defaults,
create_model_and_diffusion,
add_dict_to_argparser,
args_to_dict
)
from scdiffusionX.DiffusionBackbone import dist_util
/home/lep/miniconda3/envs/scmuldiff/lib/python3.8/site-packages/lightning_lite/__init__.py:29: DeprecationWarning: Deprecated call to `pkg_resources.declare_namespace('lightning_lite')`.
Implementing implicit namespace packages (as specified in PEP 420) is preferred to `pkg_resources.declare_namespace`. See https://setuptools.pypa.io/en/latest/references/keywords.html#keyword-namespace-packages
__import__("pkg_resources").declare_namespace(__name__)
/home/lep/miniconda3/envs/scmuldiff/lib/python3.8/site-packages/torchvision/io/image.py:13: UserWarning: Failed to load image Python extension: libtorch_cuda_cu.so: cannot open shared object file: No such file or directory
warn(f"Failed to load image Python extension: {e}")
/home/lep/miniconda3/envs/scmuldiff/lib/python3.8/site-packages/pytorch_lightning/__init__.py:45: DeprecationWarning: Deprecated call to `pkg_resources.declare_namespace('pytorch_lightning')`.
Implementing implicit namespace packages (as specified in PEP 420) is preferred to `pkg_resources.declare_namespace`. See https://setuptools.pypa.io/en/latest/references/keywords.html#keyword-namespace-packages
__import__("pkg_resources").declare_namespace(__name__)
Based on the trained model, you can calculate the attention map of the DCA. Load data, autoencoder and diffusion model:
[2]:
encoder_config = "script/training_autoencoder/configs/encoder/encoder_multimodal.yaml"
dataset_path = '/stor/lep/diffusion/multiome/openproblem_filtered.h5mu'
covariate_keys = "cell_type"
num_class = 22
ae_path = "/stor/lep/workspace/multi_diffusion/CFGen/project_folder/experiments/train_autoencoder_openproblem_multimodal/checkpoints/last.ckpt"
[3]:
mdata = mu.read_h5mu(dataset_path)
real_cell = mdata['rna'][::5].X.toarray()
real_cell2 = mdata['atac'][::5].X.toarray()
# real_cell = mdata['rna'][mdata['rna'].obs['cell_type']=='CD4+ T activated'].X.toarray()[::7]
# real_cell2 = mdata['atac'][mdata['atac'].obs['cell_type']=='CD4+ T activated'].X.toarray()[::7]
real_cell.shape
[3]:
(13850, 13431)
[4]:
# load autoencoder
with open(encoder_config, 'r') as file:
yaml_content = file.read()
autoencoder_args = yaml.safe_load(yaml_content)
# Initialize encoder
encoder_model = EncoderModel(in_dim={'atac': real_cell2.shape[1], 'rna': real_cell.shape[1]},
n_cat=mdata['rna'].obs[covariate_keys].unique().shape[0],
conditioning_covariate=covariate_keys,
encoder_type='learnt_autoencoder',
**autoencoder_args)
# Load weights
encoder_model.load_state_dict(torch.load(ae_path)["state_dict"])
[4]:
<All keys matched successfully>
[8]:
# set diffusion backbone config
defaults = dict(
clip_denoised=True,
batch_size=16,
sample_fn="ddim",
multimodal_model_path="/stor/lep/workspace/multi_diffusion/MM-Diffusion/outputs/checkpoints_cross/open_lr1e4_w512_scale124_drop0_80w_rescale10_3crossatt64_condi/model800000.pt",
output_dir="test",
classifier_scale=0,
devices='0',
is_strict=True,
all_save_num= 1024,
seed=42,
load_noise="",
data_dir=dataset_path,
condition='cell_type',
)
defaults.update(model_and_diffusion_defaults())
parser = argparse.ArgumentParser()
defaults['rna_dim'] = '1,100'
defaults['atac_dim'] = '1,100'
defaults['num_channels'] = 128
defaults['num_res_blocks'] = 1
defaults['resblock_updown'] = True
defaults['num_class'] = 22
defaults['class_cond'] = True
add_dict_to_argparser(parser, defaults)
[9]:
# load diffusion backbone
args = parser.parse_known_args()[0]
args.rna_dim = [int(i) for i in args.rna_dim.split(',')]
args.atac_dim = [int(i) for i in args.atac_dim.split(',')]
dist_util.setup_dist(args.devices)
print("creating model and diffusion...")
multimodal_model, multimodal_diffusion = create_model_and_diffusion(
**args_to_dict(args, [key for key in model_and_diffusion_defaults().keys()])
)
multimodal_model.load_state_dict_(
dist_util.load_state_dict(args.multimodal_model_path, map_location="cpu"), is_strict=args.is_strict
)
multimodal_model.to(dist_util.dev())
optimizer2 = optim.Adam(multimodal_model.parameters(), lr=0.001)
# mdata = mu.read_h5mu(args.data_dir)
from sklearn.preprocessing import LabelEncoder
labels = mdata['rna'].obs[args.condition].values
label_encoder = LabelEncoder()
label_encoder.fit(labels)
classes_all = label_encoder.transform(labels)
rna = mdata['rna']#[mdata['rna'].obs['cell_type']=='Erythroblast']
atac = mdata['atac']#[mdata['atac'].obs['cell_type']=='Erythroblast']
0
creating model and diffusion...
For different cell type, we have different cell-type specific attention maps:
[ ]:
# ordered type list
type_list = np.array(['CD14+ Mono', 'ID2-hi myeloid prog', 'CD16+ Mono', 'cDC2',
'pDC', 'HSC', 'G/M prog','Lymph prog','MK/E prog', 'Naive CD20+ B',
'B1 B', 'Transitional B', 'Plasma cell','CD4+ T naive','CD4+ T activated',
'CD8+ T', 'CD8+ T naive','NK', 'ILC', 'Proerythroblast','Erythroblast','Normoblast', ])
Since there are three DCAs in the scDiffusion-X, we can calculate the attention map in all the three DCAs. However, ablation study showed that the information richness are highest in the seconde DCA module and the last time step. Therefore, the analysis for gene regulatory will be conduct in this attention map. This will provide a stable output, and you don’t need additional experiments to select hyperparameters.
[13]:
# calculate attention maps in each layers
time_step = 1
down_sample = 2
model_kwargs = {}
batch = {}
x1 = torch.tensor(rna[::down_sample].X.toarray(),requires_grad=True)
x2 = torch.tensor(atac[::down_sample].X.toarray(),requires_grad=True)
batch["X_norm"] = {'rna':x1,'atac':x2}
z = encoder_model.encode(batch)
video_start = z['rna'].unsqueeze(1).to(dist_util.dev())
audio_start = z['atac'].unsqueeze(1).to(dist_util.dev())
model_kwargs["label"] = torch.tensor(classes_all[::down_sample]).to(dist_util.dev())
noise ={"video":torch.randn_like(video_start),\
"audio":torch.randn_like(audio_start)}
#0 means t_th step, 1 means the audio gives groundtruth, 2 means the video gives the groundtruth
# condition_index = x_start["condition"]
t = (torch.ones(video_start.shape[0], device=dist_util.dev())*time_step).to(dtype=torch.int)
video_t = multimodal_diffusion.q_sample(video_start, t, noise = noise["video"])#.detach()
audio_t = multimodal_diffusion.q_sample(audio_start, t, noise = noise["audio"])#.detach()
att_layer1 = []
att_layer1_atac = []
att_layer2 = []
att_layer2_atac = []
att_layer3 = []
att_layer3_atac = []
for celltype in type_list:
index = (rna[::down_sample].obs['cell_type'] == celltype)
# sample_id = np.random.choice(np.arange(0, index.sum()), size=22, replace=False)
video_t_i = video_t[index][:10000]#[sample_id]
audio_t_i = audio_t[index][:10000]#[sample_id]
t_i = t[index][:10000]#[sample_id]
labels = model_kwargs["label"][index][:10000]#[sample_id]
noise_pred_video, noise_pred_video, att_maps = multimodal_model(video_t_i,audio_t_i,t_i,labels,return_attvec=True)
att_layer1.append(att_maps[3])
att_layer2.append(att_maps[7])
att_layer3.append(att_maps[11])
att_layer1_atac.append(att_maps[2])
att_layer2_atac.append(att_maps[6])
att_layer3_atac.append(att_maps[10])
With the attention maps, you can find the key positions in different cell type, for example, in the CD4+ T activated cell (number 14 in all cell type):
[ ]:
# fine the key elements in gene and peak. 14 is the target cell type index (CD4+ T activated here)
plt.rcParams['pdf.fonttype'] = 42
plt.rcParams['ps.fonttype'] = 42
plt.figure(figsize=(12,12))
plt.imshow(att_layer2[14].mean(0).cpu().detach().numpy()+att_layer2_atac[14].mean(0).cpu().detach().numpy().T,vmax=0.2,cmap='coolwarm')
plt.ylabel('peak')
plt.xlabel('gene')
Text(0.5, 0, 'gene')
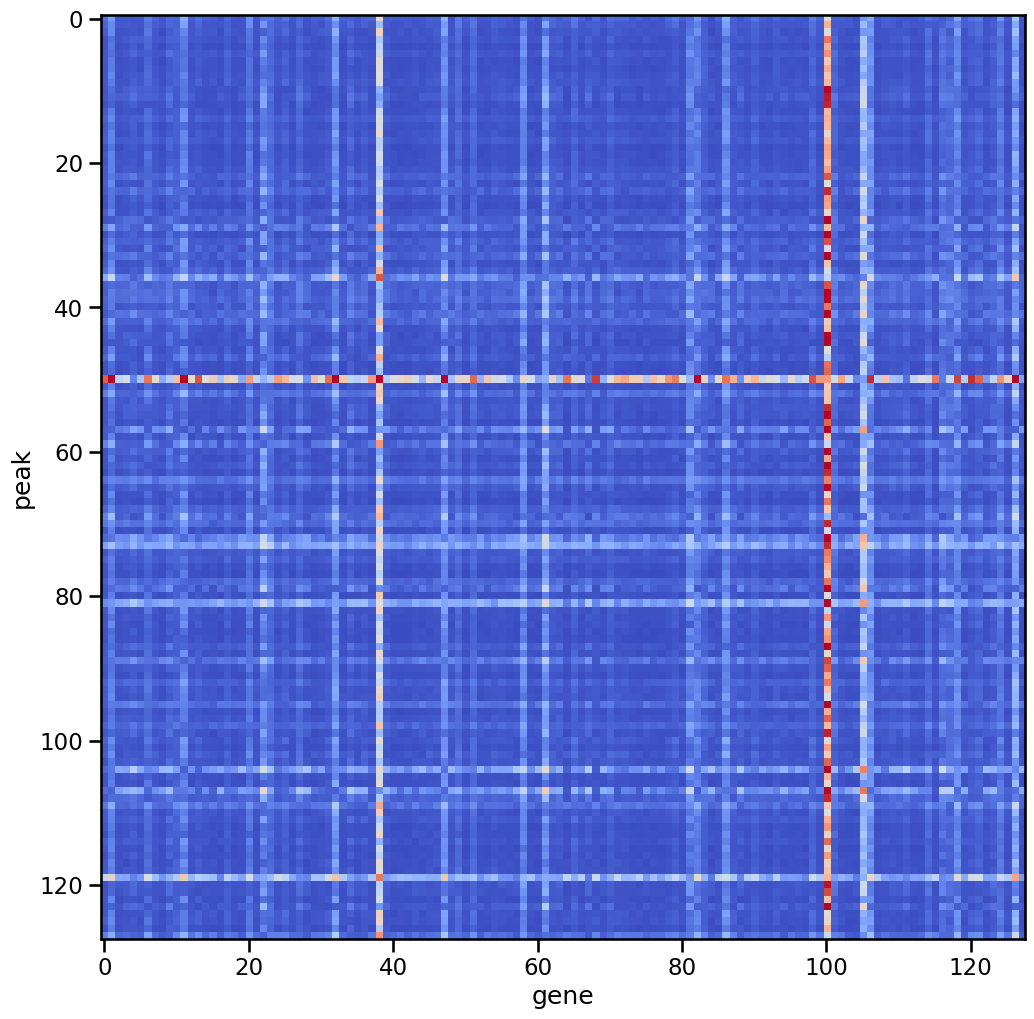
These elements are later used to obtain the cell-type specific heterogeneous gene regulatory network.
[19]:
plt.rcParams['pdf.fonttype'] = 42
plt.rcParams['ps.fonttype'] = 42
cross_map = att_layer2[14].mean(0).cpu().detach().numpy()+att_layer2_atac[14].mean(0).cpu().detach().numpy().T
flattened_indices = np.argsort(cross_map, axis=None)[-10:]
positions = np.unravel_index(flattened_indices, cross_map.shape)
max_values = cross_map[positions]
print("The position (x,y) of the top10 elements:")
print('x(peak):', positions[0])
print('y(gene):', positions[1])
print("Top 10 values:", max_values)
The position (x,y) of the top10 elements:
x(peak): [ 50 50 81 79 50 45 28 50 57 107]
y(gene): [ 11 47 100 100 38 100 100 126 100 100]
Top 10 values: [0.22636247 0.2294471 0.23520574 0.23761243 0.23839217 0.2453197
0.24915317 0.25201848 0.25299454 0.2953034 ]
Also, you can find which RNA elements are most concerned by peak. For example, in the second DCA:
[21]:
plt.rcParams['pdf.fonttype'] = 42
plt.rcParams['ps.fonttype'] = 42
key_col = np.zeros((len(att_layer2),att_layer2[0].shape[-1]))
for i in range(len(att_layer2)):
for j in range(att_layer2[i].shape[0]):
att_mean = att_layer2[i][j].mean(0).detach().cpu()
key_c = np.where(att_mean>0.35)[0]
for c in key_c:
key_col[i,c] += 1
key_col[i] = key_col[i]/att_layer2[i].shape[0]
plt.figure(figsize=(32,8))
sns.heatmap(key_col, cmap='coolwarm', vmax=0.2)
plt.yticks(ticks=np.arange(type_list.shape[0]), labels=type_list)
plt.xticks(ticks=np.arange(0,key_col.shape[1],5), labels=np.arange(0,key_col.shape[1],5))
plt.title('what RNA element to focus')
[21]:
Text(0.5, 1.0, 'what gene element to focus')
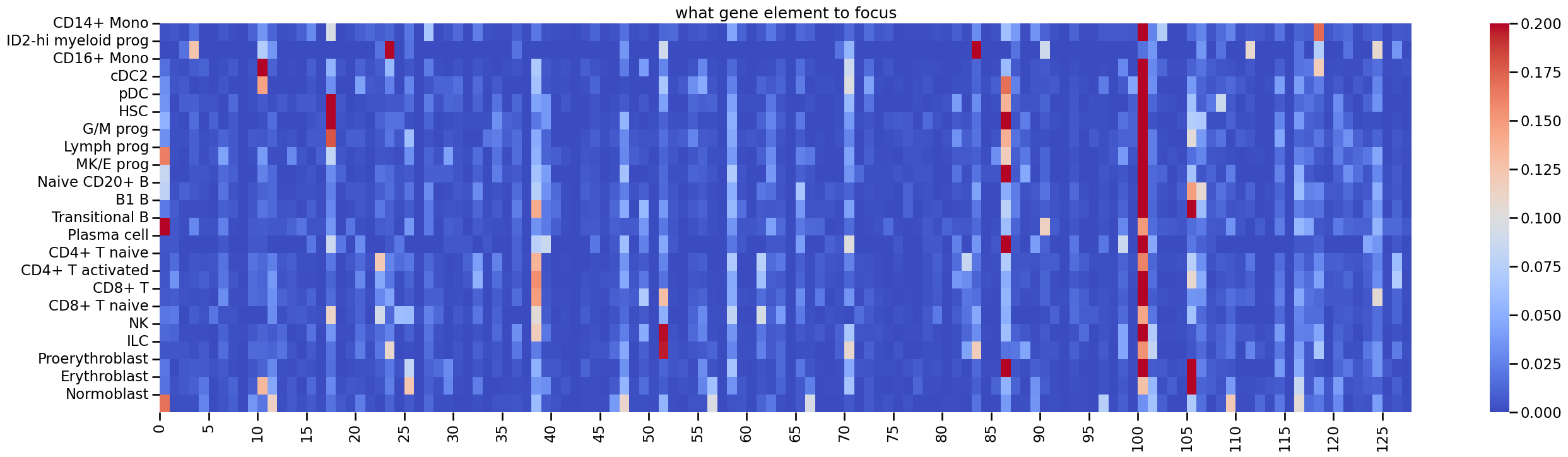
You can see apparent cell type specific pattern in this map. Similarly, you can also obtain the map about which ATAC elements are most concerned by gene:
[22]:
plt.rcParams['pdf.fonttype'] = 42
plt.rcParams['ps.fonttype'] = 42
key_col = np.zeros((len(att_layer2_atac),att_layer2_atac[0].shape[-1]))
for i in range(len(att_layer2_atac)):
for j in range(att_layer2_atac[i].shape[0]):
att_mean = att_layer2_atac[i][j].mean(0).detach().cpu()
key_c = np.where(att_mean>0.35)[0]
for c in key_c:
key_col[i,c] += 1
key_col[i] = key_col[i]/att_layer2_atac[i].shape[0]
plt.figure(figsize=(32,8))
sns.heatmap(key_col, cmap='coolwarm', vmax=0.2)
plt.yticks(ticks=np.arange(type_list.shape[0]), labels=type_list)
plt.xticks(ticks=np.arange(0,key_col.shape[1],5), labels=np.arange(0,key_col.shape[1],5))
plt.title('what ATAC elements to focus')
[22]:
Text(0.5, 1.0, 'what peak elements to focus')
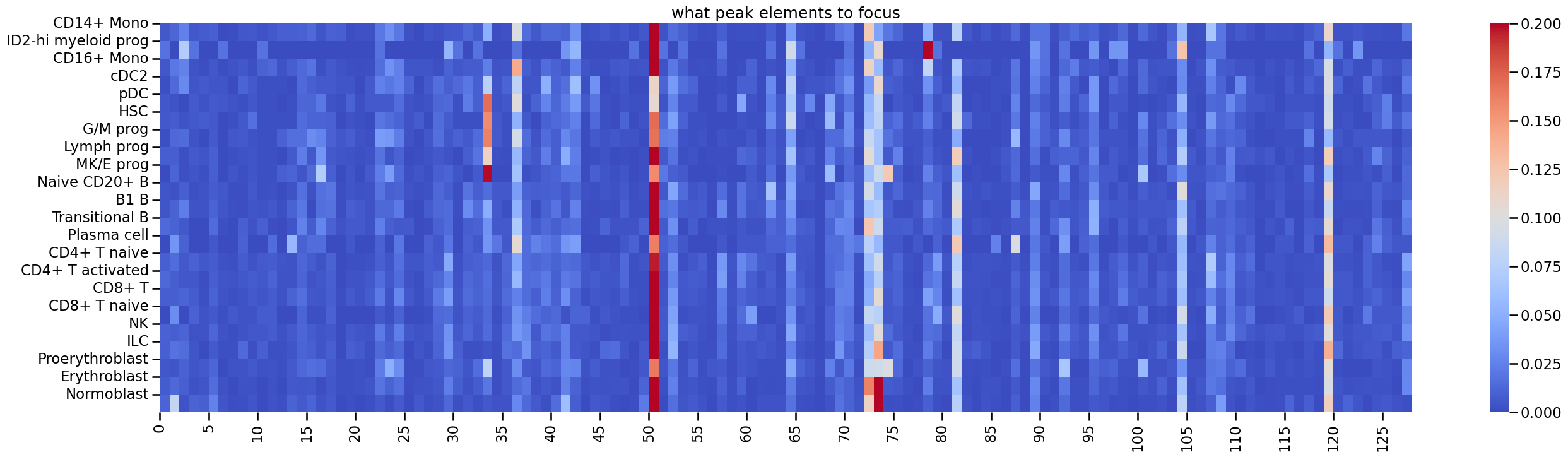
Next, we show you how to use gradient back propagation approch to analysis these attention maps. For analysis the chromosomes and genes, you need to first import the reference gene data. Here we use the hg38 reference gene from the genecode. You can download it from: https://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_47/gencode.v47.annotation.gtf.gz
[ ]:
final_df = pd.read_csv('hg38/gencode_hg38.csv')
Here we use CD4+ T activated as an example to analysis the top related genes/peaks to the cell type specific elements and construct the cell-type specific heterogeneous gene regulatory network. First, prepare the inference data (the scRNA-seq and scATAC-seq of CD4+ T activated cell):
[23]:
time_step = 1
interested_type = type_list[14]
down_sample = 5
model_kwargs = {}
batch = {}
x1 = torch.tensor(rna[::down_sample].X.toarray(),requires_grad=True)
x2 = torch.tensor(atac[::down_sample].X.toarray(),requires_grad=True)
index = list(range(rna[::down_sample].shape[0])) if interested_type=='all' else (rna[::down_sample].obs['cell_type'] == interested_type)
batch["X_norm"] = {'rna':x1,'atac':x2}
z = encoder_model.encode(batch)
video_start = z['rna'].unsqueeze(1).to(dist_util.dev())
audio_start = z['atac'].unsqueeze(1).to(dist_util.dev())
model_kwargs["label"] = torch.tensor(classes_all[::down_sample]).to(dist_util.dev())
noise ={"video":torch.randn_like(video_start),\
"audio":torch.randn_like(audio_start)}
t = (torch.ones(video_start.shape[0], device=dist_util.dev())*time_step).to(dtype=torch.int)
video_t = multimodal_diffusion.q_sample(video_start, t, noise = noise["video"])#.detach()
audio_t = multimodal_diffusion.q_sample(audio_start, t, noise = noise["audio"])#.detach()
video_t = video_t[index]
audio_t = audio_t[index]
t = t[index]
labels = model_kwargs["label"][index]
Then you can use the gradient backpropagation approch to obtain the gene/peak related to the key elements we get in the previous section. note: the returned att_vec has the following content att_vec[0]-att_vec[3] for first cross attention att_vec[0] - rna feature vector ; att_vec[1] - atac feature vector att_vec[2] - attention map of atac to rna (what peak to focus) att_vec[3] - attention map of rna to atac (what gene to focus) [4]-[7] for second cross attention, [8]-[11] for third cross attention Here we use the second cross attention as an example.
[24]:
all_key_genes = []
all_key_peaks = []
all_position = []
for key_ele in np.unique(positions[1]):
batch["X_norm"] = {'rna':x1,'atac':x2}
z = encoder_model.encode(batch)
video_start = z['rna'].unsqueeze(1).to(dist_util.dev())
audio_start = z['atac'].unsqueeze(1).to(dist_util.dev())
model_kwargs["label"] = torch.tensor(classes_all[::down_sample]).to(dist_util.dev())
noise ={"video":torch.randn_like(video_start),\
"audio":torch.randn_like(audio_start)}
t = (torch.ones(video_start.shape[0], device=dist_util.dev())*time_step).to(dtype=torch.int)
video_t = multimodal_diffusion.q_sample(video_start, t, noise = noise["video"])
audio_t = multimodal_diffusion.q_sample(audio_start, t, noise = noise["audio"])
video_t = video_t[index]
audio_t = audio_t[index]
t = t[index]
labels = model_kwargs["label"][index]
noise_pred_video, noise_pred_audio, att_vec = multimodal_model(video_t,audio_t,t,labels,return_attvec=True)
att_vec[4][:,:,key_ele].mean().backward(create_graph=True,)
top_k = mdata['rna'].shape[1]
values, indices = torch.topk(abs(x1.grad[index]).mean(0)*(x1[index].sum(0)>int(x1[index].shape[0]*0.2)), top_k)#
top_gene = np.array([mdata['rna'].var_names[id] for id in np.array(indices)])
all_key_genes += list(top_gene[:100])
multimodal_model.zero_grad()
encoder_model.zero_grad()
x1.grad.zero_()
x2.grad.zero_()
for key_ele in np.unique(positions[0]):
batch["X_norm"] = {'rna':x1,'atac':x2}
z = encoder_model.encode(batch)
video_start = z['rna'].unsqueeze(1).to(dist_util.dev())
audio_start = z['atac'].unsqueeze(1).to(dist_util.dev())
model_kwargs["label"] = torch.tensor(classes_all[::down_sample]).to(dist_util.dev())
noise ={"video":torch.randn_like(video_start),\
"audio":torch.randn_like(audio_start)}
t = (torch.ones(video_start.shape[0], device=dist_util.dev())*time_step).to(dtype=torch.int)
video_t = multimodal_diffusion.q_sample(video_start, t, noise = noise["video"])
audio_t = multimodal_diffusion.q_sample(audio_start, t, noise = noise["audio"])
video_t = video_t[index]
audio_t = audio_t[index]
t = t[index]
labels = model_kwargs["label"][index]
noise_pred_video, noise_pred_audio, att_vec = multimodal_model(video_t,audio_t,t,labels,return_attvec=True)
att_vec[5][:,:,key_ele].mean().backward(create_graph=True)
values2, indices2 = torch.topk(abs(x2.grad[index]).mean(0)*(x2[index].sum(0)>int(x2[index].shape[0]*0.05)), 36553)#
top_peak = np.array([mdata['atac'].var_names[id] for id in np.array(indices2)])
all_key_peaks += list(top_peak[:100])
multimodal_model.zero_grad()
encoder_model.zero_grad()
x1.grad.zero_()
x2.grad.zero_()
/home/lep/miniconda3/envs/scmuldiff/lib/python3.8/site-packages/torch/autograd/__init__.py:251: UserWarning: Using backward() with create_graph=True will create a reference cycle between the parameter and its gradient which can cause a memory leak. We recommend using autograd.grad when creating the graph to avoid this. If you have to use this function, make sure to reset the .grad fields of your parameters to None after use to break the cycle and avoid the leak. (Triggered internally at ../torch/csrc/autograd/engine.cpp:1171.)
Variable._execution_engine.run_backward( # Calls into the C++ engine to run the backward pass
[25]:
key_ele_gene = {}
key_ele_peak = {}
for i in range(np.unique(positions[1]).shape[0]):
key_ele_gene[np.unique(positions[1])[i]]=all_key_genes[i*100:(i+1)*100]
for i in range(np.unique(positions[0]).shape[0]):
key_ele_peak[np.unique(positions[0])[i]]=all_key_peaks[i*100:(i+1)*100]
# np.savez('hete_net_14.npz', elements=positions, key_gene=np.array([key_ele_gene], dtype=object), key_peak=np.array([key_ele_peak], dtype=object))
For each key RNA elements, we have the top related genes. For each key ATAC elements, we have the top related peaks. In this example, we have 5 distinct RNA elements and 7 distinct ATAC elements:
[26]:
key_ele_gene.keys(),key_ele_peak.keys()
[26]:
(dict_keys([11, 38, 47, 100, 126]), dict_keys([28, 45, 50, 57, 79, 81, 107]))
For each RNA element, we obtain the top 100 related genes:
[ ]:
# an example of the gene related to 47th RNA element
for gene in key_ele_gene[47]:
print(gene)
MAF
MAST4
LINC00299
FAAH2
AC139720.1
ADAM19
GZMK
PBXIP1
ARRDC3
RGS1
OST4
CD226
NOL4L
HBP1
AC022217.3
STK17A
LPXN
A2M
GALM
TAGAP
ZFYVE28
ETNK1
TRIR
UQCRB
MTRNR2L12
GNLY
ITGA6
NELL2
KLRB1
LEPROTL1
CD28
CD5
GPSM3
TNFSF8
AC013652.1
TGFBR3
TRAT1
CSGALNACT1
ZNF331
DNAJB1
EPHA4
DDX3X
MT-ND3
FRY
CFDP1
ZNF217
MYO1G
SFMBT1
SNRNP200
TSHZ2
EPSTI1
RNPC3
RNF166
RANBP2
DDX18
PTPRM
GATA3
NSRP1
CREM
UBL5
IFRD1
H1FX
CMSS1
GPR155
S100A8
DGKA
LRIG1
PCSK7
LBH
CD2AP
EIF4E3
LAT
GBP5
LPIN2
PRKCQ-AS1
IGF2R
TESPA1
TIAL1
GPR174
PCMTD1
TOB1
CAPN2
TRG-AS1
ATP5MG
RORA-AS1
WWP2
TRAF3IP3
FAM53B
GOLGA8A
LINC-PINT
FCMR
NLRC3
NOP58
GIMAP4
MAPRE2
KLRG1
COMMD6
CHD3
MT-ATP6
EIF3M
For each ATCA element, we obtain the top 100 related peaks:
[ ]:
# an example to fine the related peaks' corresponding (nearst) gene to 104th ATAC element
for peak in key_ele_peak[104]:
print(find_nearest_gene(final_df, peak)[0][0])
GZMA
SMG6
GGA3
C4orf50
DPP4
ENSG00000309832
SCAMP2
SH3RF3
ENSG00000308252
ELAPOR2
COPS8-DT
STAM
ENSG00000301728
ENSG00000250602
ENSG00000309950
ITK
CCDC7
SLC7A6
LINC02646
ENSG00000298044
ENSG00000259704
ENSG00000285635
ITGB1-DT
DAB1
PRKCB
ENSG00000289377
RNA5SP431
TSPEAR
ZDHHC11B
CRACDL
MED15
ARHGAP15
ENSG00000299620
RN7SKP166
NLRC5
ENSG00000233538
RUNX2
NPM1P2
DSTN
NDFIP2-AS1
CXCR5
PVT1
RNF216
ADAM19
SNX9
ENSG00000300850
SUPT3H
ENSG00000291013
HDAC4
LRRC8C-DT
SPOCK2
ENSG00000299987
PRDM2
XYLT1
CFAP299
PECAM1
ARHGAP10
EMILIN2
CNNM2
LRRC8C
BEGAIN
LEF1
CDK5RAP1
EVI5
DGKZ
ENSG00000309104
FIRRM
EHD4
OXNAD1
ENSG00000288724
CORO1B
ARHGAP25
TIAM1
ADARB1
ENSG00000225649
BTF3L4P3
PRKCA
KCNJ1
USP12
SUSD4
ENSG00000308933
BEX5
ENSG00000306277
ITK
TRAF1
TRAF2
Y_RNA
ENSG00000288729
IKZF1
BPGM
LINC01991
ZBTB46
FYB1
NIBAN1
TMEM106B
ENSG00000300042
ETS1
EPAS1
GATA3
SLC25A26
You can also print all the related gene/peak for this specific cell type.
[116]:
for name in np.unique(all_key_genes):
print(name)
A2M
AC013652.1
AC022217.3
AC079793.1
AC139720.1
AC245297.3
ADAM19
AGFG1
AL136456.1
ANKRD36
ANO6
ANTXR2
ANXA11
ANXA6
AP001011.1
APBA2
AQR
ARHGAP45
ARHGEF6
ARHGEF7
ARL4C
ARRDC3
ATF2
ATF7IP2
ATP10A
ATP5IF1
ATP5MG
BRD1
BRD7
BTBD11
C12orf57
CAPN2
CBFB
CCL5
CCND2
CCSER2
CD226
CD28
CD2AP
CD37
CD3D
CD3E
CD3G
CD48
CD5
CD81
CDC42SE1
CDK5RAP2
CDKN1B
CDV3
CFDP1
CHD3
CHURC1
CLIC1
CLIP1
CMSS1
CNOT1
COMMD6
COX6A1
COX6B1
CREM
CRIP1
CSGALNACT1
CYTOR
DAZAP2
DDX10
DDX18
DDX3X
DDX46
DENND4C
DGKA
DIPK1A
DNAJB1
DUSP16
EDF1
EIF3M
EIF4E3
EIF5B
ELP2
EPHA4
EPSTI1
ERAP2
ERP29
ETNK1
FAAH2
FAM53B
FARS2
FCMR
FLNA
FMN1
FRY
GADD45B
GALM
GATA3
GBP5
GIMAP4
GNLY
GOLGA8A
GOLGA8B
GPR155
GPR174
GPR183
GPSM3
GRAP2
GZMA
GZMK
H1FX
HBP1
HELB
HINT1
HIVEP1
HMGN1
IFRD1
IGF2R
IKZF2
IL10RA
IL2RG
ITGA6
IVNS1ABP
KAT2B
KLRB1
KLRG1
LAT
LBH
LDHB
LEF1
LEPROTL1
LINC-PINT
LINC00299
LINC00513
LINC00861
LINC01138
LINC02694
LPIN2
LPXN
LRIG1
LTB
MAF
MALT1
MAPRE2
MAST4
MCL1
MED23
MT-ATP6
MT-CO1
MT-CO2
MT-CYB
MT-ND1
MT-ND2
MT-ND3
MT-ND4
MT-ND5
MTAP
MTFR1
MTRNR2L12
MYO1F
MYO1G
NABP1
NCOA7
NELL2
NKG7
NLRC3
NME2
NOL4L
NOP58
NR1D2
NR2C2
NSRP1
NT5DC1
ODF2L
OPTN
OST4
P2RY8
PARP14
PBXIP1
PCAT1
PCMTD1
PCNX2
PCSK7
PIK3CD
PIK3IP1
PLCG2
PPP1R16B
PPP1R2
PPP2R2A
PRDM1
PRKCQ-AS1
PSMB8
PSMB9
PSME1
PSME2
PTPN2
PTPRM
RANBP2
RASGRF2
RBM5
RCAN3
RELL1
RETREG1
RGS1
RNF166
RNMT
RNPC3
RORA-AS1
S100A11
S100A8
SCML4
SELENOF
SELL
SERINC1
SERP1
SET
SFI1
SFMBT1
SLC39A10
SLC9A3R1
SNRNP200
SP140
SPAG9
SPN
SPOCK2
SSR4
STAM
STAT1
STK17A
STRN
SYTL2
TAF15
TAGAP
TBCA
TCF25
TCF7
TESPA1
TGFBR3
TIAL1
TMEM117
TMEM135
TMEM245
TMEM63A
TMEM65
TNFSF8
TOB1
TPR
TRAC
TRAF3IP3
TRAT1
TRG-AS1
TRIR
TSHZ2
TTC39B
TTN
UBASH3B
UBL3
UBL5
UQCRB
USP36
VAMP2
WWP2
XAF1
XRN2
ZFYVE28
ZNF217
ZNF331
ZNF652
[56]:
for name in np.unique(all_key_peaks):
print(name.replace('-', ':', 1))
chr1:100400124-100401025
chr1:100452357-100453271
chr1:100927083-100927969
chr1:101357133-101358041
chr1:108703358-108704087
chr1:116556331-116557223
chr1:116738599-116739461
chr1:116877748-116878647
chr1:117658424-117659293
chr1:13702529-13703436
chr1:151830384-151831295
chr1:154791005-154791840
chr1:156124729-156125582
chr1:160664488-160665308
chr1:167441913-167442794
chr1:169694194-169695012
chr1:181159372-181160283
chr1:184843493-184844395
chr1:18948169-18949079
chr1:19074232-19075107
chr1:193478979-193479887
chr1:194289309-194290212
chr1:19687950-19688899
chr1:198187853-198188770
chr1:198668796-198669696
chr1:203682362-203683265
chr1:206715677-206716563
chr1:206736878-206737712
chr1:206784197-206785074
chr1:206816648-206817552
chr1:221712956-221713713
chr1:223363861-223364743
chr1:226444671-226445586
chr1:227773894-227774801
chr1:229253318-229254119
chr1:232923711-232924616
chr1:234730807-234731559
chr1:234758605-234759500
chr1:25796521-25797356
chr1:38977438-38978360
chr1:39183542-39184421
chr1:53413067-53413977
chr1:57307135-57308052
chr1:58941500-58942417
chr1:6460071-6460918
chr1:66351635-66352527
chr1:7948785-7949679
chr1:89126913-89127800
chr1:89610395-89611174
chr10:100433498-100434398
chr10:101782956-101783855
chr10:101829240-101829859
chr10:103053822-103054756
chr10:11150311-11151057
chr10:114544018-114544904
chr10:118698201-118699103
chr10:130301012-130301843
chr10:132159023-132159874
chr10:132616039-132616954
chr10:14586608-14587451
chr10:15370768-15371634
chr10:17648142-17649017
chr10:32459052-32459968
chr10:3887605-3888502
chr10:6129697-6130570
chr10:62082566-62083336
chr10:62383858-62384706
chr10:70578097-70578970
chr10:71742058-71742855
chr10:72083674-72084561
chr10:7269110-7270002
chr10:74586019-74586373
chr10:8056783-8057586
chr10:8059858-8060669
chr10:8183013-8183936
chr10:8404471-8405371
chr10:86400922-86401559
chr10:94545287-94546198
chr11:118001419-118002330
chr11:118046000-118046868
chr11:118342298-118343130
chr11:118343914-118344801
chr11:118398579-118399462
chr11:118434144-118434988
chr11:118610635-118611510
chr11:118892199-118893106
chr11:118895347-118896208
chr11:118919417-118920270
chr11:122696361-122697108
chr11:128422365-128423266
chr11:128716647-128717362
chr11:128830021-128830745
chr11:13923934-13924707
chr11:2467054-2467936
chr11:326708-327585
chr11:34248219-34249123
chr11:46330951-46331771
chr11:60912006-60912871
chr11:61083380-61084214
chr11:64900008-64900880
chr11:64971626-64972512
chr11:67446050-67446955
chr11:75507926-75508817
chr11:76231850-76232766
chr12:107027316-107028219
chr12:107371952-107372863
chr12:111993327-111994151
chr12:116924319-116925194
chr12:121092552-121093446
chr12:121534276-121535160
chr12:122244509-122245416
chr12:12481948-12482804
chr12:31890392-31891274
chr12:3947489-3948397
chr12:44876103-44876888
chr12:4506601-4507502
chr12:51323273-51323660
chr12:53236155-53237061
chr12:538972-539840
chr12:56160236-56161112
chr12:56338407-56339298
chr12:64679315-64680211
chr12:66287887-66288774
chr12:67483017-67483937
chr12:89397905-89398807
chr12:92398393-92399288
chr12:96377579-96378476
chr13:24264721-24265634
chr13:27130907-27131808
chr13:30773927-30774845
chr13:31806834-31807754
chr13:48185968-48186783
chr13:51801825-51802554
chr13:79451289-79452183
chr13:95185606-95186491
chr13:99215207-99216079
chr14:100078718-100079623
chr14:100546109-100547024
chr14:104077727-104078624
chr14:104864868-104865700
chr14:105859561-105860473
chr14:106004697-106005601
chr14:22536675-22537546
chr14:31123960-31124845
chr14:35203636-35204566
chr14:61335637-61336537
chr14:61337534-61338420
chr14:61384998-61385913
chr14:61452226-61453106
chr14:63650099-63651010
chr14:64673449-64674333
chr14:64696175-64697090
chr14:65846857-65847697
chr14:68779666-68780441
chr14:76929703-76930618
chr14:91078165-91079067
chr14:91867136-91868005
chr14:91869727-91870613
chr14:96126149-96127037
chr14:97977855-97978638
chr14:98223498-98224398
chr14:99191607-99192440
chr14:99260703-99261509
chr15:38683347-38684186
chr15:39580097-39580829
chr15:41969730-41970635
chr15:52256052-52256956
chr15:59803412-59804326
chr15:60398635-60399362
chr15:60580335-60581247
chr15:63841693-63842596
chr15:65828530-65829164
chr15:74845020-74845932
chr15:76996523-76997407
chr15:84736221-84737026
chr15:90376632-90377526
chr15:90407754-90408659
chr15:90856419-90857303
chr15:91469644-91470553
chr16:17396593-17397446
chr16:23948592-23949371
chr16:3076727-3077596
chr16:4465692-4466564
chr16:56194073-56194976
chr16:56610178-56610882
chr16:56938561-56939407
chr16:56999154-57000041
chr16:68275506-68276415
chr16:69443812-69444760
chr16:75074944-75075846
chr16:75098258-75099121
chr16:79271899-79272807
chr16:79602009-79602814
chr16:82655145-82656015
chr16:89841000-89841926
chr17:14201341-14202069
chr17:2222723-2223644
chr17:22520971-22521869
chr17:35177260-35178134
chr17:35890485-35891389
chr17:40068518-40069395
chr17:41341185-41342069
chr17:416545-417324
chr17:47706303-47707218
chr17:48453244-48454167
chr17:4900630-4901435
chr17:50779702-50780594
chr17:50871081-50872000
chr17:55238079-55238959
chr17:58152757-58153665
chr17:64081812-64082735
chr17:64347855-64348761
chr17:65173229-65174076
chr17:66246065-66246982
chr17:66673418-66674321
chr17:7045502-7046284
chr17:74279837-74280755
chr17:75242186-75243068
chr17:76191615-76192480
chr17:76772777-76773624
chr17:78381460-78382383
chr18:24011234-24012138
chr18:24118716-24119593
chr18:26215356-26216249
chr18:2889538-2890462
chr18:3305005-3305907
chr18:3604370-3605286
chr18:45962302-45963107
chr18:48842355-48843221
chr18:49820035-49820856
chr18:63199549-63200482
chr18:74493455-74494332
chr18:75070749-75071674
chr18:76720971-76721851
chr18:77071623-77072497
chr19:10142504-10143403
chr19:1192089-1192994
chr19:12289016-12289922
chr19:14523130-14523809
chr19:1874446-1875202
chr19:29667597-29668460
chr19:38559276-38560183
chr19:45016286-45016813
chr19:49568873-49569604
chr19:55626136-55627020
chr19:6674614-6675507
chr19:8575296-8576160
chr2:100643842-100644708
chr2:10493327-10494224
chr2:109143915-109144670
chr2:112749531-112750371
chr2:12856642-12857557
chr2:130824130-130825045
chr2:134180968-134181886
chr2:136281174-136282058
chr2:143258158-143258944
chr2:148524266-148525160
chr2:162011419-162012322
chr2:162073705-162074539
chr2:169107472-169108345
chr2:172461730-172462575
chr2:203719732-203720625
chr2:203740776-203741641
chr2:231425702-231426623
chr2:234463766-234464686
chr2:236967748-236968648
chr2:237434666-237435561
chr2:239321738-239322604
chr2:239622889-239623785
chr2:25285669-25286568
chr2:3222587-3223541
chr2:37585897-37586767
chr2:38653667-38654458
chr2:46351481-46352389
chr2:64213982-64214908
chr2:68721296-68722201
chr2:70068771-70069672
chr2:8303267-8304167
chr2:84782862-84783699
chr2:86825785-86826701
chr2:97718315-97719218
chr2:9781923-9782825
chr2:98835648-98836483
chr20:17610356-17611190
chr20:32509392-32510287
chr20:33369086-33369899
chr20:33370186-33370967
chr20:37014887-37015797
chr20:44070094-44070957
chr20:56460219-56461107
chr20:5835644-5836501
chr20:59165899-59166780
chr20:59222182-59223084
chr20:63178907-63179770
chr20:63750718-63751637
chr20:63781778-63782643
chr20:63955405-63955932
chr21:31132619-31133502
chr21:31354364-31355279
chr21:33936260-33937107
chr21:42238240-42239050
chr21:44653848-44654712
chr21:45224215-45225137
chr21:45266271-45267160
chr22:20549212-20550082
chr22:21632241-21633147
chr22:23242818-23243716
chr22:28822719-28823582
chr22:31278752-31279658
chr22:36161912-36162824
chr22:37604694-37605549
chr22:38984754-38985657
chr22:39099183-39100060
chr22:39909947-39910781
chr22:40055220-40056072
chr22:42269289-42269888
chr3:101518102-101519030
chr3:10242083-10242987
chr3:10263571-10264487
chr3:108806105-108807005
chr3:114231457-114232317
chr3:129118563-129119428
chr3:13880365-13880913
chr3:141410965-141411664
chr3:15054778-15055708
chr3:157130164-157131026
chr3:161310665-161311584
chr3:16311689-16312606
chr3:183469071-183469980
chr3:187999683-188000493
chr3:189324432-189325342
chr3:195609055-195609975
chr3:20103982-20104869
chr3:42555977-42556867
chr3:45693811-45694740
chr3:45869842-45870757
chr3:45950537-45951393
chr3:45976431-45977306
chr3:46284878-46285709
chr3:47056983-47057848
chr3:5019292-5020111
chr3:50316440-50317375
chr3:52892769-52893536
chr3:66306733-66307635
chr3:93470137-93471048
chr3:98564403-98565236
chr4:101327919-101328850
chr4:101526945-101527852
chr4:102518496-102519380
chr4:108166451-108167274
chr4:108168395-108169302
chr4:109648320-109649227
chr4:139266376-139267288
chr4:142404922-142405822
chr4:147799720-147800598
chr4:150827927-150828830
chr4:151408604-151409471
chr4:153114512-153115286
chr4:153521906-153522746
chr4:36322690-36323595
chr4:38130982-38131752
chr4:40283758-40284639
chr4:42671222-42672116
chr4:6180666-6181567
chr4:80500345-80501257
chr4:88382379-88383277
chr4:9603857-9604711
chr4:98706006-98706881
chr5:126508586-126509494
chr5:132055860-132056795
chr5:134086651-134087565
chr5:134100138-134101004
chr5:134145581-134146492
chr5:141691799-141692663
chr5:142104258-142105158
chr5:143434663-143434978
chr5:157159108-157159910
chr5:157193466-157194380
chr5:157540575-157541445
chr5:157603140-157603999
chr5:175895419-175896307
chr5:176529558-176530397
chr5:177370464-177371383
chr5:32531559-32532298
chr5:35854069-35854835
chr5:35924615-35925514
chr5:50428281-50429186
chr5:50711017-50711902
chr5:55102204-55103009
chr5:758890-759748
chr5:76325627-76326526
chr5:79237150-79238038
chr5:82356835-82357723
chr5:912209-913120
chr5:96537124-96538040
chr5:96876010-96876900
chr5:99037505-99038423
chr6:106195248-106196022
chr6:106360242-106361056
chr6:111605414-111606171
chr6:118796318-118797197
chr6:128520232-128521095
chr6:130513458-130514370
chr6:134240448-134241323
chr6:135128441-135129193
chr6:137589166-137590037
chr6:137717209-137718081
chr6:137819559-137820441
chr6:137883369-137884137
chr6:139160942-139161695
chr6:149494664-149495516
chr6:152184645-152185578
chr6:156396154-156396981
chr6:157761019-157761927
chr6:161076050-161076976
chr6:16420687-16421610
chr6:16481408-16482302
chr6:16654449-16655357
chr6:167119411-167120299
chr6:25041903-25042661
chr6:25865748-25866655
chr6:33856388-33857297
chr6:44932553-44933350
chr6:45586350-45587183
chr6:46011193-46012082
chr6:5173974-5174864
chr6:90233702-90234616
chr6:90397741-90398585
chr7:102671306-102672199
chr7:130929981-130930880
chr7:131007235-131008109
chr7:134669562-134670439
chr7:138875579-138876491
chr7:143408550-143409388
chr7:148628361-148629251
chr7:151206760-151207615
chr7:151429554-151430468
chr7:157612889-157613741
chr7:25028420-25029301
chr7:25862097-25863026
chr7:36285968-36286801
chr7:43669145-43670048
chr7:44257555-44258478
chr7:44914809-44915687
chr7:50321377-50322290
chr7:5144108-5144958
chr7:5641673-5642553
chr7:76008727-76009631
chr7:7944453-7945383
chr7:831969-832857
chr7:86978549-86979448
chr7:96285350-96286266
chr7:98400722-98401593
chr8:100409994-100410784
chr8:11820105-11820996
chr8:127981094-127981953
chr8:130244631-130245519
chr8:133062292-133063207
chr8:133520098-133520965
chr8:140790627-140791490
chr8:143549172-143549998
chr8:144262129-144262975
chr8:19496774-19497670
chr8:19510850-19511750
chr8:2130737-2131641
chr8:2133645-2134527
chr8:23219299-23219951
chr8:38359162-38360012
chr8:38362558-38363375
chr8:38368244-38369121
chr8:60796326-60797227
chr8:66467545-66468455
chr8:66490502-66491419
chr8:7354769-7355690
chr8:80138400-80139278
chr8:80864470-80865384
chr8:9011665-9012579
chr8:94119723-94120662
chr9:105405231-105406120
chr9:120903772-120904680
chr9:127955141-127956054
chr9:131709682-131710612
chr9:132581744-132582654
chr9:133371066-133371858
chr9:135978758-135979682
chr9:136902182-136902928
chr9:33446082-33446817
chr9:37528882-37529735
chr9:37531465-37532388
chr9:447376-448282
chr9:89743198-89744076
chr9:92953855-92954737
chrX:102155558-102156374
chrX:110229540-110230457
chrX:119682023-119682946
chrX:130339422-130340343
chrX:136586094-136587006
chrX:44344209-44344794
chrX:49053241-49054181
chrX:49273048-49273948
chrX:72105154-72106048
chrX:78516874-78517789
Next, we show how to find the potential regulatory relationship between the top related genes and peaks. Again, take CD4+ T activated as an example.
[14]:
type_list
[14]:
array(['CD14+ Mono', 'ID2-hi myeloid prog', 'CD16+ Mono', 'cDC2', 'pDC',
'HSC', 'G/M prog', 'Lymph prog', 'MK/E prog', 'Naive CD20+ B',
'B1 B', 'Transitional B', 'Plasma cell', 'CD4+ T naive',
'CD4+ T activated', 'CD8+ T', 'CD8+ T naive', 'NK', 'ILC',
'Proerythroblast', 'Erythroblast', 'Normoblast'], dtype='<U19')
You need to first load the encode H3K27ac data to find the potential enhancer position.
[32]:
type_index = [14]
bed_file = []
for n in type_index:
if n in [13,14]:
bed_file.append(pd.read_csv('encode/CD4.bed', sep='\t', header=None, names=['chrom', 'start', 'end', 'name', 'score', 'strand', 'signalValue','pValue','qValue','peak']))
elif n in [1]:
bed_file.append(pd.read_csv('encode/myeloid.bed', sep='\t', header=None, names=['chrom', 'start', 'end', 'name', 'score', 'strand', 'signalValue','pValue','qValue','peak']))
elif n in [9,10,11]:
bed_file.append(pd.read_csv('encode/B cell.bed', sep='\t', header=None, names=['chrom', 'start', 'end', 'name', 'score', 'strand', 'signalValue','pValue','qValue','peak']))
elif n in [15,16]:
bed_file.append(pd.read_csv('encode/CD8.bed', sep='\t', header=None, names=['chrom', 'start', 'end', 'name', 'score', 'strand', 'signalValue','pValue','qValue','peak']))
elif n in [17]:
bed_file.append(pd.read_csv('encode/NK.bed', sep='\t', header=None, names=['chrom', 'start', 'end', 'name', 'score', 'strand', 'signalValue','pValue','qValue','peak']))
elif n in [0]:
bed_file.append(pd.read_csv('encode/CD14.bed', sep='\t', header=None, names=['chrom', 'start', 'end', 'name', 'score', 'strand', 'signalValue','pValue','qValue','peak']))
Then, calcualte the overlap of above top peaks to genes’ enhancer position or promoter (TSS) position.
[34]:
all_overlaps = []
all_peaks = []
all_tss = []
num_genes = []
num_enhancers = []
for ii, k in enumerate(type_index):
cross_map = att_layer2[k].mean(0).cpu().detach().numpy()+att_layer2_atac[k].mean(0).cpu().detach().numpy().T
flattened_indices = np.argsort(cross_map, axis=None)[-15:]
positions = np.unravel_index(flattened_indices, cross_map.shape)
max_values = cross_map[positions]
time_step = 1
interested_type = type_list[k]
down_sample = 5
model_kwargs = {}
batch = {}
x1 = torch.tensor(rna[::down_sample].X.toarray(),requires_grad=True)
x2 = torch.tensor(atac[::down_sample].X.toarray(),requires_grad=True)
index = list(range(rna[::down_sample].shape[0])) if interested_type=='all' else (rna[::down_sample].obs['cell_type'] == interested_type)
all_key_genes = []
all_key_peaks = []
all_position = []
for key_ele in np.unique(positions[1][-15:]):
batch["X_norm"] = {'rna':x1,'atac':x2}
z = encoder_model.encode(batch)
video_start = z['rna'].unsqueeze(1).to(dist_util.dev())
audio_start = z['atac'].unsqueeze(1).to(dist_util.dev())
model_kwargs["label"] = torch.tensor(classes_all[::down_sample]).to(dist_util.dev())
noise ={"video":torch.randn_like(video_start),\
"audio":torch.randn_like(audio_start)}
t = (torch.ones(video_start.shape[0], device=dist_util.dev())*time_step).to(dtype=torch.int)
video_t = multimodal_diffusion.q_sample(video_start, t, noise = noise["video"])
audio_t = multimodal_diffusion.q_sample(audio_start, t, noise = noise["audio"])
video_t = video_t[index]
audio_t = audio_t[index]
t = t[index]
labels = model_kwargs["label"][index]
noise_pred_video, noise_pred_audio, att_vec = multimodal_model(video_t,audio_t,t,labels,return_attvec=True)
att_vec[4][:,:,key_ele].mean().backward(create_graph=True,)
top_k = mdata['rna'].shape[1]
values, indices = torch.topk(abs(x1.grad[index]).mean(0)*(x1[index].sum(0)>int(x1[index].shape[0]*0.2)), top_k)#
top_gene = np.array([mdata['rna'].var_names[id] for id in np.array(indices)])
all_key_genes += list(top_gene[:100])
multimodal_model.zero_grad()
encoder_model.zero_grad()
x1.grad.zero_()
x2.grad.zero_()
for key_ele in np.unique(positions[0][-15:]):
batch["X_norm"] = {'rna':x1,'atac':x2}
z = encoder_model.encode(batch)
video_start = z['rna'].unsqueeze(1).to(dist_util.dev())
audio_start = z['atac'].unsqueeze(1).to(dist_util.dev())
model_kwargs["label"] = torch.tensor(classes_all[::down_sample]).to(dist_util.dev())
noise ={"video":torch.randn_like(video_start),\
"audio":torch.randn_like(audio_start)}
t = (torch.ones(video_start.shape[0], device=dist_util.dev())*time_step).to(dtype=torch.int)
video_t = multimodal_diffusion.q_sample(video_start, t, noise = noise["video"])
audio_t = multimodal_diffusion.q_sample(audio_start, t, noise = noise["audio"])
video_t = video_t[index]
audio_t = audio_t[index]
t = t[index]
labels = model_kwargs["label"][index]
noise_pred_video, noise_pred_audio, att_vec = multimodal_model(video_t,audio_t,t,labels,return_attvec=True)
att_vec[5][:,:,key_ele].mean().backward(create_graph=True)
values2, indices2 = torch.topk(abs(x2.grad[index]).mean(0)*(x2[index].sum(0)>int(x2[index].shape[0]*0.1)), mdata['atac'].shape[1])#
top_peak = np.array([mdata['atac'].var_names[id] for id in np.array(indices2)])
all_key_peaks += list(top_peak[:200])
multimodal_model.zero_grad()
encoder_model.zero_grad()
x1.grad.zero_()
x2.grad.zero_()
chromosome_segments = np.unique(all_key_peaks)
final_df['seqname'] = [name.split('_')[0] for name in final_df['seqname']]
enhancers = []
for segment in chromosome_segments:
overlaps = find_enhancer_overlaps(bed_file[ii], segment)
if not overlaps.empty:
for i in range(overlaps.shape[0]):
enhancers.append('-'.join([overlaps['chrom'].values[i],str(overlaps['start'].values[i]),str(overlaps['end'].values[i])]))
overlap_gene = []
overlap_seg = []
# 查找每个染色体片段的最近基因
for segment in chromosome_segments:
gene_name, feature = find_nearest_gene(segment)
if gene_name is not None:
overlap_gene+=gene_name
overlap_seg.append(segment)
results = []
for segment in chromosome_segments:
result = check_tss_overlap(final_df, segment)
if len(result)>0:
results.append(result)
all_tss.append(np.unique(np.concatenate(results)[np.in1d(np.concatenate(results),np.unique(all_key_genes))]))
all_overlaps.append(np.array(overlap_gene)[np.in1d(overlap_gene,np.unique(all_key_genes))])
all_peaks.append(np.array(chromosome_segments))
num_genes.append(np.unique(all_key_genes).shape[0])
num_enhancers.append(len(enhancers))
num_overlap = np.array(overlap_gene)[np.in1d(overlap_gene,np.unique(all_key_genes))].shape[0]
num_tts = np.unique(np.concatenate(results)[np.in1d(np.concatenate(results),np.unique(all_key_genes))]).shape[0]
print(f'{type_list[k]}: {np.unique(all_key_genes).shape[0]} genes, {chromosome_segments.shape[0]} peaks, {len(enhancers)} enhancer, {num_overlap} overlap, {num_tts} tts,' )
CD4+ T activated: 291 genes, 537 peaks, 545 enhancer, 31 overlap, 6 tts,
Then, you can further obtain the top related peaks that overlaped with the TSS and the potential enhancer of the top related genes. Here we need the loop data between enhancers and TSS, these loop data can be found in HiChip.
[36]:
def check_overlap(segment1, segment2):
"""check if two segments overlaped"""
chrom1, start1, end1 = parse_segment(segment1)
chrom2, start2, end2 = parse_segment(segment2)
return chrom1 == chrom2 and start1 <= end2 and end1 >= start2
def find_overlapping_segments(list1, list2):
"""find overlaped segments in two segments list"""
overlapping = []
for seg1 in list1:
for seg2 in list2:
if check_overlap(seg1, seg2):
overlapping.append(seg1)
return overlapping
type_index_loop = [14]
bed_file_loop = []
for n in type_index_loop:
if n in [13,14,15,16]:
bed_file_loop.append(pd.read_csv('hichip/loop_info_T_cell_hichipper.csv',))
elif n in [9,10,11]:
bed_file_loop.append(pd.read_csv('hichip/loop_info_B_cell_hichipper.csv',))
for iid, item in enumerate([0]): # 在type_index中的位置
print(type_list[type_index_loop[iid]])
print('all overlap: ',all_overlaps[item])
print('tss: ')
for tss in all_tss[item]:
print(tss)
chrom = final_df[(final_df['gene_name']==tss) & (final_df['feature']=='gene')][['seqname','start','end']].values[0][0]
start_id = str(final_df[(final_df['gene_name']==tss) & (final_df['feature']=='gene')][['seqname','start','end']].values[0][1])[:2]
gene_seg = final_df[(final_df['gene_name']==tss) & (final_df['feature']=='gene')][['seqname','start','end','strand']].values[0]
if gene_seg[-1] == '+':
tss_seg = '-'.join([gene_seg[0],str(gene_seg[1]-3000),str(gene_seg[1])])
else:
tss_seg = '-'.join([gene_seg[0],str(gene_seg[2]),str(gene_seg[2]+3000)])
print(tss_seg)
overlaps = find_enhancer_overlaps_loop(bed_file_loop[iid], tss_seg)
if overlaps.empty:
continue
enhancers = []
for kkk in range(overlaps.shape[0]):
enhancers.append('-'.join([overlaps['chrom'].values[kkk],str(int(overlaps['start'].values[kkk])),str(int(overlaps['end'].values[kkk]))]))
enhancers.append('-'.join([overlaps['chrom'].values[kkk],str(int(overlaps['start2'].values[kkk])),str(int(overlaps['end2'].values[kkk]))]))
selected_peak = [it for it in all_peaks[item] if it.startswith(chrom+'-'+start_id)]
overlaps_enhancer = find_overlapping_segments(selected_peak, enhancers)
print('overlaps_enhancer: ',np.unique(overlaps_enhancer))
CD4+ T activated
all overlap: ['SELL' 'TRAF3IP3' 'TGFBR3' 'TGFBR3' 'GATA3' 'CD3D' 'CD5' 'DUSP16'
'DUSP16' 'TESPA1' 'KLRG1' 'FMN1' 'USP36' 'LPIN2' 'MALT1' 'CD28' 'EPHA4'
'GALM' 'NOL4L' 'SFMBT1' 'TCF7' 'RETREG1' 'PRDM1' 'SCML4' 'LINC-PINT'
'LINC00513' 'STK17A' 'CSGALNACT1' 'CSGALNACT1' 'TNFSF8' 'P2RY8']
tss:
CD28
chr2-203703517-203706517
overlaps_enhancer: ['chr2-203706134-203707025']
CD5
chr11-61099489-61102489
overlaps_enhancer: ['chr11-61021436-61022320' 'chr11-61041942-61042854'
'chr11-61101933-61102835']
DIPK1A
chr1-92961522-92964522
overlaps_enhancer: ['chr1-92962295-92963080']
FMN1
chr15-33194714-33197714
overlaps_enhancer: []
RETREG1
chr5-16617101-16620101
overlaps_enhancer: ['chr5-16616577-16617478']
TESPA1
chr12-54984762-54987762
overlaps_enhancer: ['chr12-54984440-54985280']